




Hepatitis B is considered an important public health problem worldwide because it is a chronic infection with a risk factor for cirrhosis and cellular hepatocellular carcinoma. In Brazil, the Rondônia State ranks first in the Northern region regarding the number of deaths due to hepatitis B. In the Amazon basin, genotype F is considered specific to the Americas identified in native populations. But few data on HBV genotyping and phylogenetic analysis are available. The objective of this study was to evaluate the genotypes and subgenotypes of the hepatitis B virus in indigenous people chronic carriers residing in cities of Guajará Mirim and Nova Mamoré in state of Rondônia/Brazil, on the border with Bolivia. A fragment of 417 bp (S gene) was amplified by PCR and submitted to nucleotide sequencing. The genotypes and subgenotypes of the HBV strains were determined through phylogenetic inference using genomic sequences from 197 representatives of the genotypes (A-H). Of the 41 chronic hepatitis B patients enrolled in this study, 27 were HBV-DNA positive. Of the 27 DNA-HBV positives, 39% (17/41) had individual HBV infection and 27% (10/41) were coinfected with HDV. The frequency of genotypes was 40.7% (11/27) for genotype D (HBV-D), 33.3% (9/27) for genotype F (HBV-F) and 25.9% (7/27) for genotype A (HBV-A) with circulating subgenotypes F2, F4, D2, D3, A1, and A2. We characterized the genotypes and subgenotypes of HBV circulating among in indigenous in the State of Rondônia shows for the first time the HBV/D genotype whit greater frequency circulating in nativos of state Rondônia. In conclusion, our findings showed a diversity of HBV genotypes, which is also found in other Brazilian geographical regions.
The hepatitis B virus (HBV) is responsible for acute and chronic in liver infection. HBV infection has a high impact on public health worldwide1 due to the potential risk of progression to liver cirrhosis and hepatocellular carcinoma (HCC).2,3 The HBV genome is made up of partially double DNA strand with approximately 3,200 base pairs (bp) organized into four transcription units or open reading frames (ORFs), known as P (polymerase), S (surfaces), C (core), and X (protein X).4,5
The HBV replication strategy is preceded by a reverse transcription step; in this step, the lack of polymerase review activity produces high rates of nucleotide substitutions,6 which allow classification into 10 genotypes of A through J and 24 subgenotypes (A1-A3, B1-B5, C1-C6, D1-D6 and F1-F4).7,8 The geographic distribution of genotypes and subgenotypes is related to the origin of the isolates.9 In Brazil, genotypes A (HBV-A), D (HBV-D) and F (HBV-F) are predominant, which reflects the miscegenation of the Brazilian population.10-12
The western Amazon basin is considered an endemic area for HBV infection13-16 with a low to high prevalence distribution.17-19 In the Brazilian occidental Amazon, a significant rate of HBV infection in indigenous peoples was discovered in the state of Rondônia, which borders Bolivia.16
Rondônia state in northern Brazil is known for having the second highest number of cases of hepatitis B,19 poor access to health services, risky cultural habits, and a migratory movement of Indigenous people between villages, all favoring the endemicity of HBV infection and hepatitis Delta virus (HDV) coinfection. The viral characteristics of HBV (genotypes, viral load, specific mutations) and coinfection with HDV are determinants of the course of infection, but are unknown in the region.3,20-23
Therefore, the present study has the main objective of evaluating genotypic and subgenotypic diversity and observing the presence of mutations in the HBV S gene in chronic carriers infected and coinfected with circulating HDV in the Indigenous population living in the border region between Brazil and Bolivia in the western Brazilian Amazon.
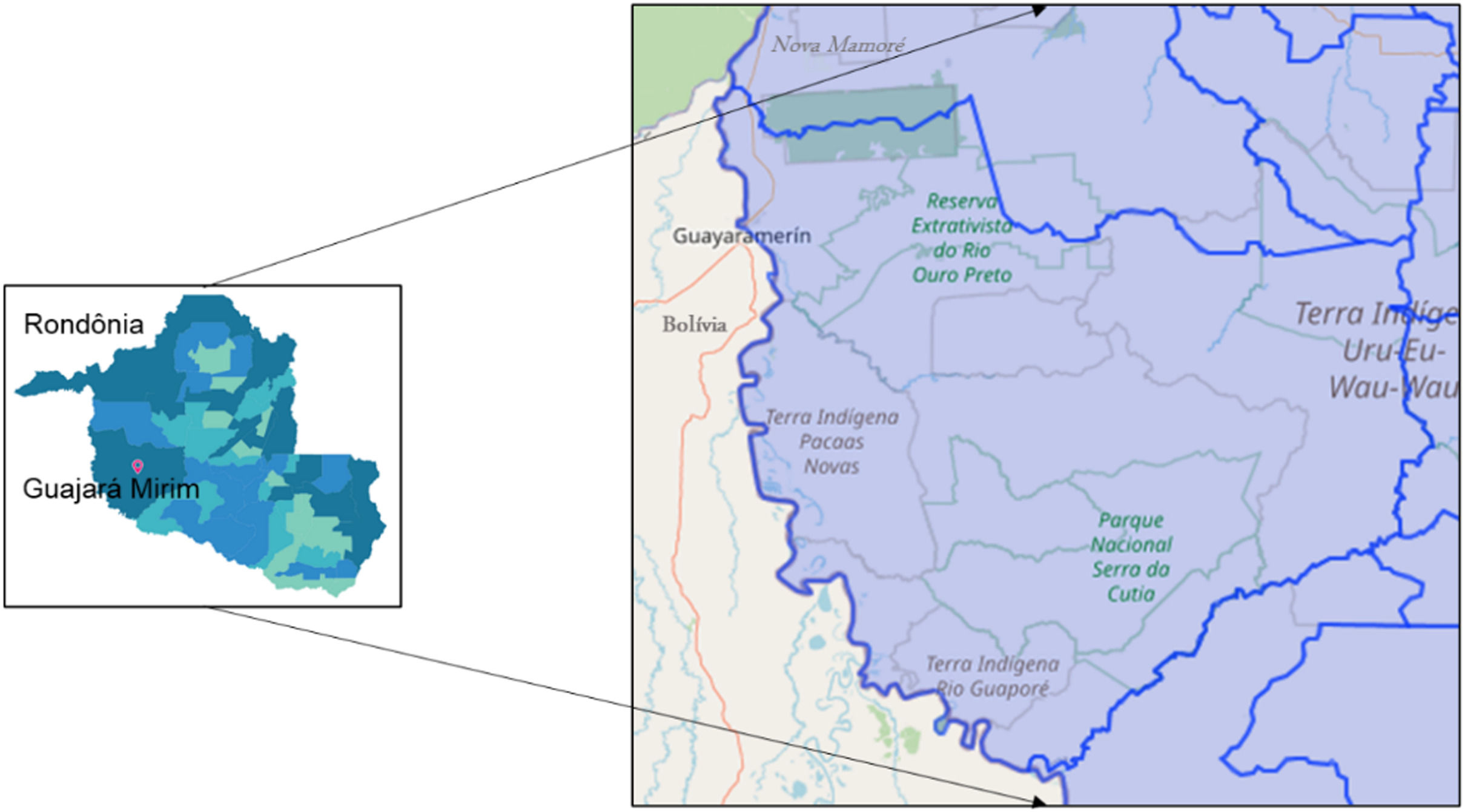
Materials and methodsThe study area encompasses the municipalities of Guajará Mirim and Nova Mamoré, state of Rondônia/Brazil, on the border of Guayaramerin/Beni Bolivia (Fig. 1). The study was accompanied by a clinical and laboratory outpatient reference center for Viral Hepatitis, which is located at the Tropical Medicine Research Center - CEPEM. DNA-HBV molecular tests were performed by the research team at FIOCRUZ-RO Molecular Virology Laboratory.
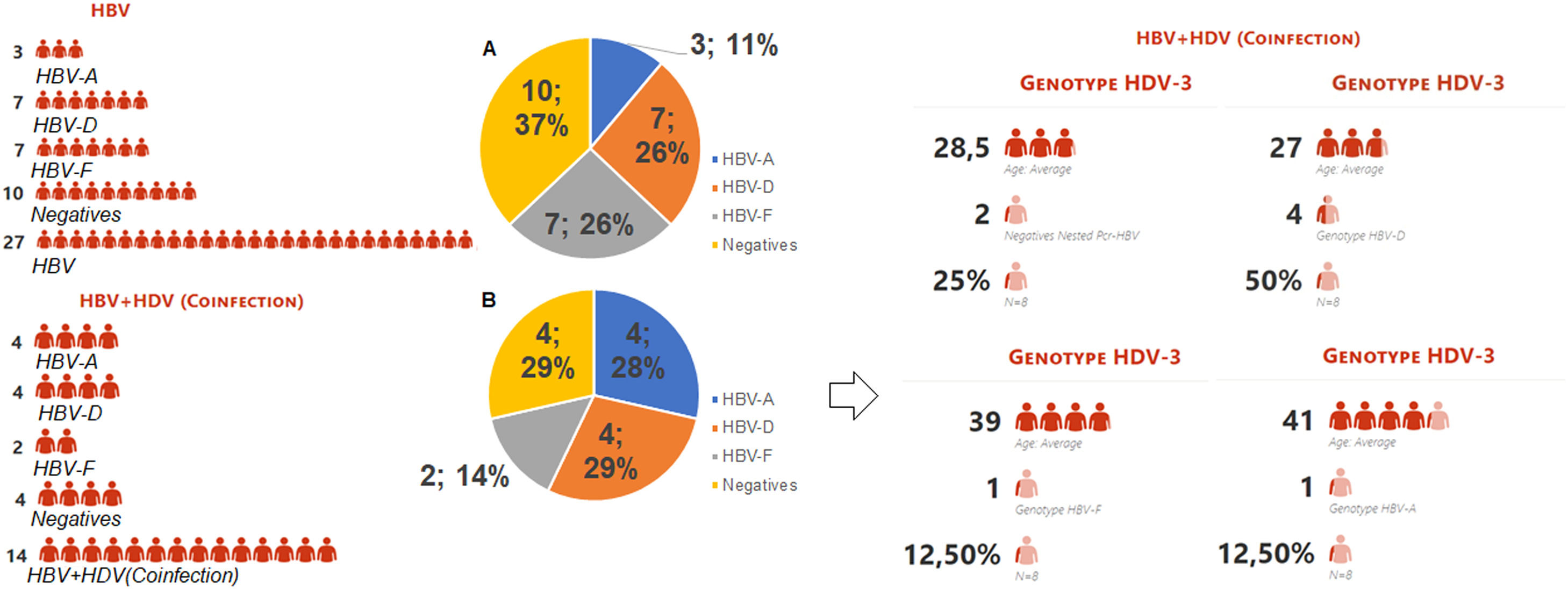
Representation of type infection HBV stratified by genotypes. Legend: HBV-individual infection with hepatitis B; HBV+HDV- coinfection between HBV and HDV viruses; HBV-A (genotype A); HBV-D (genotype D); HBV-F (genotype D); Graph A: Represents the 27 indigenous individuals with individual HBV infection, 10 (37%) of which are DNA-HBV negative, 17 DNA-HBV positive characterized in genotypes HBV-A (11%), HBV-D (26%) and HBV- F (26%). Graph B: Represents the 14 indigenous people coinfected with HDV, 4 (29%) being DNA-HBV negative, 10 DNA-HBV positives characterized in genotypes HBV-A (28%), HBV-D (29%) and HBV-F (14%). Among the coinfected individuals, 8 were RNA-HDV positive, characterized by the genotype HDV-3, were coinfected with 25% DNA-HBV negative and 75% DNA-HBV positive, and were characterized as HDV-A (12.50%), HDV-D (50%), and HDV-F (12.50%). The mean age among those coinfected with genotype D was 27 years, 28.5 years among the DNA-HBV negative in nested PCR, 39 years for HBV-F and 41 years among those coinfected with HBV-A.
The sample size of this study represents 22% Of the total of 187 HBsAg reactive indigenous people registered in the DSEI (Special Indigenous Sanitary District),16 41 (22%) patients were included in this study based on availability of sociodemographic data. Chronic hepatitis B was defined as persistence of HBsAg for more than six months, presence of total anti-HBc and HBsAg in individuals of both sexes aged 18 years or older. The samples were collected at the Central Laboratory of the State of Rondônia (LACEN/RO) during a blood test for viral evaluation. Participants from to the nine ethnicities denominated Oro Mon, Oro Nao, Canoé, Macurap, Aruá, Oro At, Oro Waram Xinjein, Cujubim and Jabuti were included.
Hepatitis Delta serology for was performed at the Central Laboratory of Rondônia (LACEN), in accordance to the Ministry of Health's technical guidelines for the diagnosis of viral hepatitis.
The study was approved by the National Council of Ethics in Research – CONEP, with opinion number 1.718,840 in collaboration with the District Council of Indigenous Health – CONDISI and the Special Sanitary District of Indigenous – DSEI Porto Velho.
Extraction and Nested-PCR of DNA-HBVPositive samples on qualitative HBV PCR were selected for molecular analysis, in which DNA was isolated from 200 µL of patient serum using the QIAamp Viral DNA Mini Kit (©ฏQIAGEN) according to the manufacturer's instructions and eluted in 60 µL. HBV DNA amplification was performed by nested PCR using type-specific primers7 for the target region. The first-round PCR primers amplify a 447 bp fragment of the S region with primers FHBS1 (position 244 to 267; 5’-GAGTCTAGACTCGTGGTGGACTTC-3’) and RHBS1 (position 688 to 691; 5’ GCTAAATKGCACTAG TAAACTGAGCCA-3’), and the second-round primers amplify a 417-bp fragment with primers FHBS2 (position 255 to 278; 5’-CGTGGTGGACTTCTCTCAATTTTC-3’) and RHBS2 (position 648 to 671; 5’-GCCARGAGAAAC GGRCTGAGGCCC-3’).
HBV genotyping and subgenotypingHBV genotypes and subgenotypes of the strains were determined through phylogenetic inference using genomic sequences from 197 representatives of the genotypes (A-H) and a sequence of HBV from ducks – DHBV (JX469898). The subgenotypic classification used in this study was based on the new proposal for reclassification of HBV variants established by Yin et al.9 Then, the sequences obtained in the study were aligned under the MUSCLE algorithm in MEGA7 - Molecular Evolutionary Genetic Analysis software, using the evolutionary model General Time Reversible with Gamma distribution and Invariant Sites (GTR+G+I) chosen through the Model Analysis tool attached to MEGA7. The evolutionary analysis was performed based on Bayesian inference using Monte Carlo Markov chain (MCMC) algorithms implemented in the BEAST v.1.10.4 package.24 The evolutionary profile was estimated using the Bayesian Skyline coalescent model, relaxed molecular clock uncorrelated with log normal distribution and under a chain length of 1 × 10^8, with collection every 10,000 generations. In addition, the tree likelihood parameter was used in Tracer v.1.7.1 software (effective sample size – ESS value > 200). The Tree Annotator v.10.1 program was used to summarize the posterior distribution of the tree with 10% burn-in, and FigTree v1.4.3 was used to view/customize the annotated MCC (maximum clade credibility) tree.
HBV mutation analysisThe electropherogram produced in the sequencing using the FHBS2 and RHBS2 primers was compared with manually produced consensus sequences for mutation analysis using MEGA7 software.25 Nucleotide sequences were aligned using the MUSCLE algorithm. Mutation analysis was performed at the nucleotide and amino acid levels, as described in the literature of alterations in the S gene.
Molecular analysis of HDV coinfectionTo determine the HDV genotype, viral RNA was extracted from 200 μL of serum using the commercial QIAamp Viral RNA Mini Kit (Qiagen, Germany) according to the manufacturer's instructions. The precipitated RNA was resuspended in 50 µl of elution buffer. Then, to produce HDV cDNA, a reverse transcriptase-RT reaction was prepared using 15 μL of viral RNA and M-MLV enzyme (Moloney murine leukemia virus) (Sigma Aldrich, Saint Louis, MO, USA) at a concentration of 200 U. Both RT and PCR procedures for the detection of HDV followed the procedures described by Botelho-Souza et al. including primers, cycle conditions, nucleotide sequencing, and phylogenetic analysis.20
Statistical analysis consisted of a descriptive analysis using online software Copyright© 2021 Minitab, LLC.
ResultsEpidemiological profile of Indigenous people with HBVIn the study, 41 indigenous individuals with chronic HBV infection were included; 49% (20/41) had a family history of HBV infection, 10% (4/41) did not, and 41% (17/41) did not have this information in the record.
The average length of HBV infection among the study participants was 12.6 years (range: two to 24 years). Twelve of the 27 (45%) males in the study had been vaccinated. Vaccinated women had the longest infection period (14.57 years).
The majority (59%; n = 24) of included indigenous individuals were in the age group of less than 40 years, and there were more men (66%; n = 27) than women (34%; n = 14). Males had higher nested-PCR positivity than females (Table 1).
Clinical, virological, and demographic characteristics of HBV patients stratified by type of infection.
| Chronic carriers of HBV (n = 41) | ||||
|---|---|---|---|---|
| Male Nested-PCR + | Female Nested-PCR + | |||
| Variables | Status: Infection (n = 17) | Status: Coinfection HDV (n = 10) | Status: Infection (n = 10) | Status: Coinfection HDV (n = 4) |
| Sex | ||||
| M | 12 | 6 | 6 | 3 |
| F | 5 | 4 | 4 | 1 |
| Age | ||||
| < 40 years | 10 | 8 | 8 | 3 |
| ≥ 40 years | 7 | 2 | 2 | 1 |
| Village | ||||
| Baía da Coca | 3 | 0 | 1 | 0 |
| Baía das Onças | 0 | 2 | 0 | 0 |
| Capoeirinha | 0 | 1 | 0 | 0 |
| Casa do índio | 1 | 1 | 0 | 0 |
| Deolinda | 0 | 2 | 1 | 0 |
| Graças Deus | 0 | 0 | 1 | 0 |
| Lage Novo | 1 | 0 | 0 | 0 |
| Linha 31 | 0 | 0 | 1 | 0 |
| Ribeirão | 0 | 0 | 1 | 0 |
| Ricardo Franco | 1 | 1 | 2 | 2 |
| Rio Negro Ocaia | 3 | 0 | 1 | 0 |
| Sagarana | 4 | 0 | 1 | 0 |
| Sotério | 1 | 2 | 0 | 1 |
| Tanajura | 2 | 2 | 0 | 1 |
| Tribo Canoé | 0 | 0 | 1 | 0 |
| HBV length of Infection | ||||
| ≥ 10 years | 11 | 9 | 8 | 4 |
| < 10 years | 5 | 1 | 2 | 0 |
| Vaccinated | ||||
| Yes | 9 | 1 | 6 | 3 |
| No | 7 | 10 | 4 | 1 |
| Signs of advanced liver disease | ||||
| Hepatic cirrhosis | * | 6 | * | * |
| Hepatic cirrhosis and parenchyma Hepatic nodular | * | 1 | * | 1 |
| Hepatic cirrhosis and portal hypertension- Splenomegaly | * | 1 | * | * |
| Hepatic-heterogeneous parenchyma and portal hypertension-Splenomegaly | * | * | * | 1 |
| Hepatic-nodular parenchyma and portal hypertension-Splenomegaly | * | 1 | * | * |
| HBV Viral load | ||||
| Detectable (> 2,000UI/mL) | 9 | 1 | 2 | 0 |
| Not detectable | 7 | 10 | 6 | 4 |
| Not done | 0 | 0 | 2 | 0 |
| HDV Viral load | ||||
| Detectable | 0 | 7 | 0 | 1 |
| Not detectable | 0 | 1 | 0 | 2 |
| Not done | 17 | 3 | 10 | 1 |
Of the 41 indigenous participants in the study, 27 had a history of individual HBV infection, 14 were coinfected with HDV (Table 1), and 11 (27%) individuals were infected with the HBV-D genotype. Genotype D carriers had, on average, a longer duration of infection (13 years of infection), followed by genotypes F and A.
Forty-one percent (11/27) of phylogenetically analyzed sequences were classified as HBV-D, 33% (9/27) as HBV-F, and 26% (7/27) as HBV-A (Fig. 3). Genotype D was identified in different regions of the study site, including the village of Lage Novo, Rio Negro Ocaia, Copeirinha, Deolinda, Tanajura, Baía da Coca, Casa do índio and Sotério. Out of the F genotype obtained sequences, 15% were from individuals located in Sagarana village and adjacent riverside villages (Sotério, Rio Negro Ocaia, Baía da Coca, Ricardo Franco). Genotype A was observed in patients from Sagarana village, Rio Negro Ocaia, Deolinda, Tanajura, and Baía das Onças.
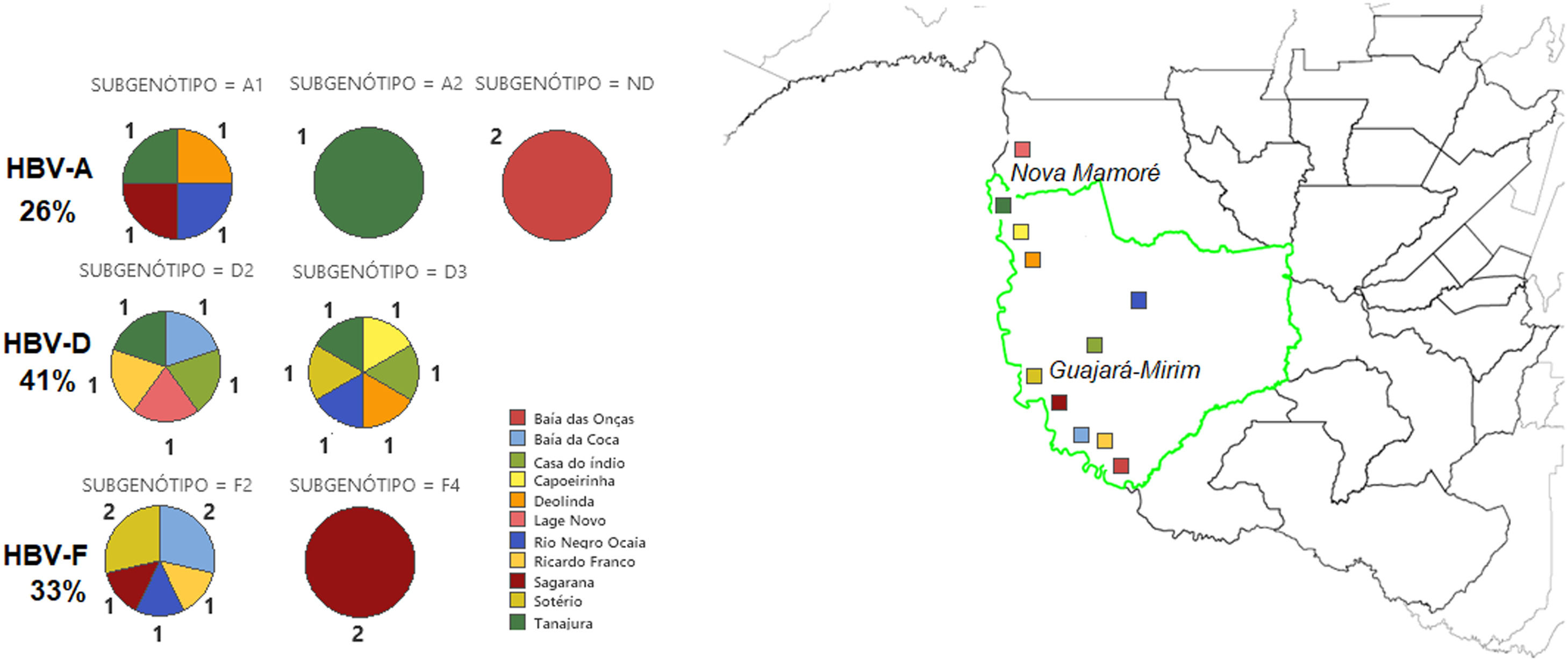
Among the HBV-D genotype sequences, five samples were HBV-D2 subgenotype and six HBV-D3 (formerly HBV-D3 and D6). Among those belonging to the HBV-F genotype, seven were confirmed as HBV-F2 subgenotypes and two HBV-F4. When observing the HBV-A clades, something different was observed; of the seven identified sequences, four were grouped in the HBV-A1 cluster, one in the HBV-A2 cluster (formerly HBV-A2 and A6), while two formed a monophyletic cluster distant from other known HBV-A subgenotypes, with 100% posterior support (Fig. 4).
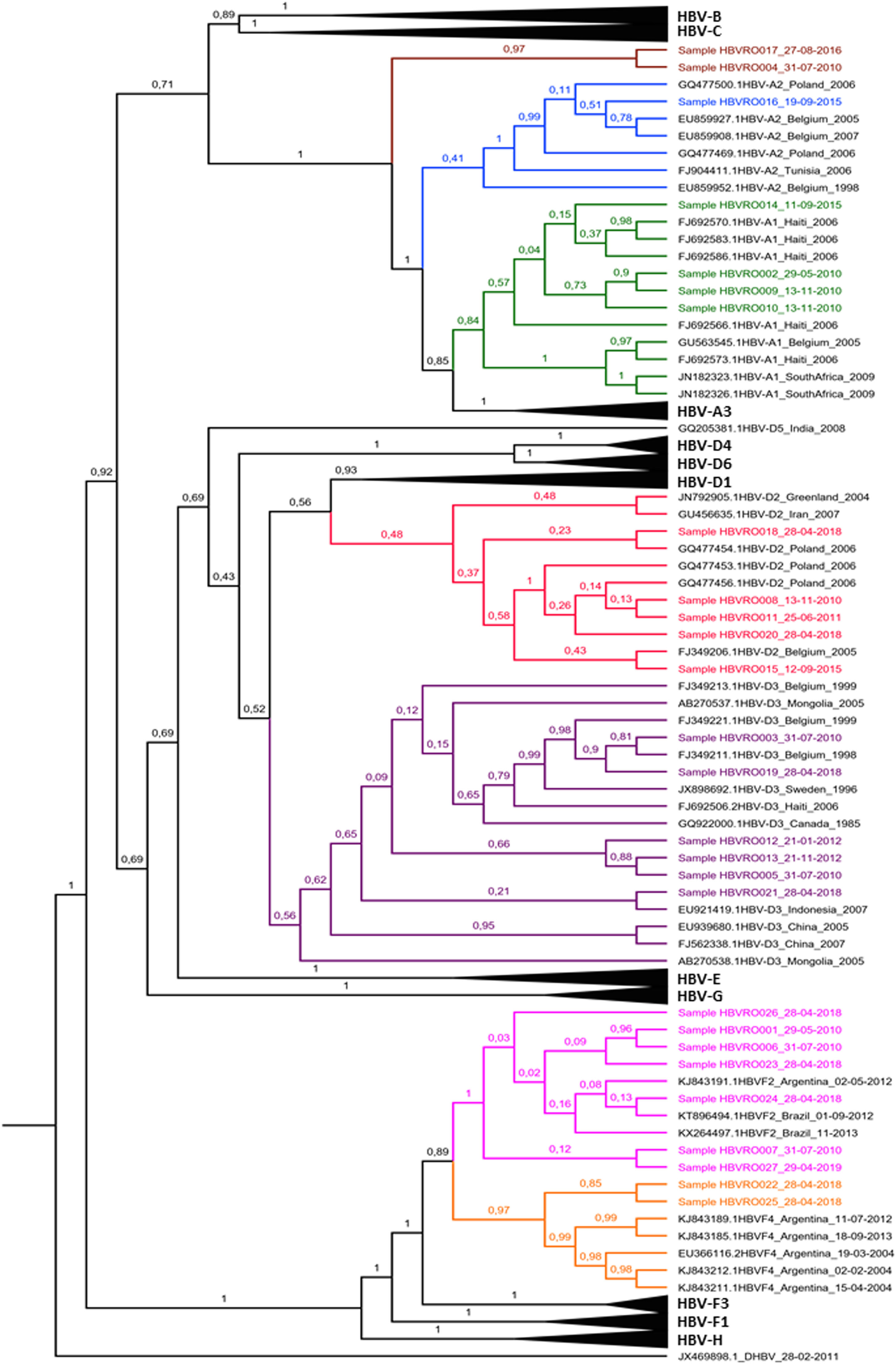
Bayesian Phylogenetic Tree. Legend: Clades whose branches do not include samples have been collapsed (black triangles). The samples are identified with 'Sample' and colored according to the clade to which they belonged: light green to A1; blue to A2; red to D2; purple to D3; pink to F2; orange to F4. The brown clade corresponds to indigenous HBV-A samples that lined up in a single cluster, unlike any cluster of the A genotype. The HBV-D sequence (JX469898) is arranged as the root of the tree. In each node, the percentage in decimal of the posterior support of these nodes is shown.
The observed mutations were more common in HBV-D and HBV-F sequences. Immune escape-related substitutions were identified (Table 2), and V106A, G112E, and T118 V/A substitutions, known to impair virus assembly, were noted.
Vaccine escape-related mutations arising in the HBsAg antigenic region.
| Identification | Escape Mutation gene S | |||||
|---|---|---|---|---|---|---|
| Sequences | 120 L | 120T | 122K | 128 V | 131N | 144A |
| HBVRO1026IND | * | * | * | V | * | * |
| HBVRO1027IND | * | T | * | * | * | * |
| HBVRO1178IND | * | T | * | * | * | * |
| HBVRO1831IND | L | * | * | * | * | * |
| HBVRO2126IND | * | * | * | * | N | * |
| HBVRO2548IND | * | * | K | * | * | * |
| HBVRO2707IND | * | * | * | * | * | A |
| HBVRO3085IND | * | * | * | V | * | * |
| HBVRO359IND | * | T | * | * | * | * |
| HBVRO4058IND | * | * | * | V | * | * |
| Total | 1 | 3 | 1 | 3 | 1 | 1 |
The vaccine escape mutant D144A was observed in the HBV-F genotype, and a sequence of indigenous coinfected with HDV was found with stop codon C69* mutation.
DiscussionHBV molecular surveillance in indigenous populations remains scarce in the region, although genotypic diversity of HBV has already been observed in indigenous and non-indigenous populations in northern Brazil.14,26-28
It was observed in this study that native HBV carriers had a time of infection that ranged from two to 24 years. It is noteworthy that among chronic carriers 49% (20/41) had at least one member of their family infected with HBV. However, due to unavailability of infection information among 41% (17/41) of participants, analyses on the risk of intrafamilial transmission were limited. A study in Brazil's southern region found a similar frequency of HBV infection among family members,29 corroborating the findings in this population.
Regarding sex, there were more chronic male patients (66%; n = 41) than females (34%; n = 41), with most participants being individuals less than 40 years old (71%; n = 29). It was observed that unvaccinated individuals had longer duration of infection than vaccinated individuals (Table 2). Among the 27 male indigenous people, only 12 were vaccinated; that is, 45% (n = 27) had an average of 11 years of HBV infection. The longer duration of infection (14.57 years) among unvaccinated female indigenous people may reflect the low vaccination coverage at the beginning of the 21st century, when indigenous populations had difficult access to villages thus not having access to health care, reflected by the low adherence to the vaccine schedule against hepatitis B.
The molecular characterization of genotypes can reveal the viral origin resulting from the historical process in populations in some regions. The HBV-F genotype is native to America, as it was frequently observed in samples from indigenous populations of Latin America (Argentina, Brazil, Bolivia, Venezuela, and Colombia) and is considered an indigenous genotype.10,26,30-32 However, in the region of the present study, bordering Brazil and Bolivia, a different genotypic pattern was observed, in which the phylogenetic evaluation classified the HBV-D genotype in 41% (11/27), with HBV-genotype F (33%) being less frequent in this population.
HBV-F was classified into two subgenotypes, HBV-F2 with 77.7% (7/9) and HBV-F4 with 23.3% (2/9). In Brazil, HBV-F2 is considered native,10 while the HBV-F4 subgenotype, despite having been identified in Brazil, has a greater distribution between the countries of Argentina and Bolivia.12,32,34,35 The presence of these subgenotypes in the region may be due to the colonization process of the region, marked by an intense migratory flow. The movement of indigenous people between riverine villages might be a factor favoring virus spread at the border, given that the sequences of the HBV-F2 and F4 subgenotypes identified were from indigenous people residing in the riverine villages Baía da Coca, Sotério, Sagarana, Rio Negro Ocaia and Ricardo Franco, located on the border with Bolivia.
In Brazil, the genotypic diversity of HBV is closely linked to the process of population and historical displacement in the region where these peoples reside. According to Mello et al.33 miscegenation in recent years was a determining factor for the genotypic distribution of HBV. This diversity of genotypes has been more evident in native populations, as these peoples have greater contact with other populations,27,33 which represents a characteristic of the Wari peoples since the process of population36 in the state of Rondônia. In Argentina, the predominance of four different HBV subgenotypes circulating in a native population was demonstrated for the first time, the Mbyá-Guarani (F4, F1, A2, and D3), from which the historical process of displacement to Brazil was related.37 Regarding genotypic evaluation, it was not possible to infer whether the Wari population has been infected by non-F genotypes for a long time or whether transmission occurred in recent times, as the size of the fragment obtained through the PCR technique was not sufficient for this type of inference, even though the fragment obtained belongs to a specific region for determining HBV genotypes and subgenotypes.
In the present study, the HBV-D genotype sequences were classified into HBV-D2 and HBV-D3 subgenotypes (formerly HBV-D3 and D6). HBV-D has been suggested as a genotype of European origin38-40 and represents approximately 24.4% of HBV variants in the Amazon.10 Spitz et al.41 suggested that HBV-D3 subgenotype sequences isolated in the Brazilian Amazon were introduced in the country several times because they are not related to European sequences. Unlike other strains, the HBV-D3 subgenotype characterized in other regions of Brazil is related to European sequences.42 However, Spitz et al.,43 in a detailed assessment of the evolutionary history of HBV-D in the Americas through different time scales, based on spatiotemporal reconstruction analyses, suggested time from the most recent common ancestor to the coincident HBV-D3 subgenotype with different migratory movements. Our isolates had a meaningful relationship with European HBV-D3, although most formed a specific cluster between sequences from Canada and Indonesia (Fig. 3). The idea of introducing these subgenotypes still in a period of migratory movements suggested by Spitz et al.43 is reinforced when we observe the findings of the present study based on the historical context of the region and the phylogenetic relationship of our isolates with sequences of European origin and non-European origin.
When evaluating the presence of mutations identified in HBV gene S, it was found that four sequences were from indigenous people with occult infection, and they had mutations V106A, I110, P120T, T118A, and A128 V. Salpini et al.44 observed that the P120T and T118A substitutions correlated with HBV-DNA reactivation in immunosuppressed patients and their absence in chronic carriers, concluding that chronic patients with such mutations are a minority. In the present study, it was not possible to determine whether these mutations were generated from cccDNA immunosuppressive therapy due to the size of the sequence generated for genomic analyses.
In contrast, in the present study, mutations P120T and T118A were observed in chronic carriers, and only one of the four individuals with occult HBV infection was treated. Study refers to position P120 as critical point for prediction of cirrhosis. Another mutation found at position C69 was significantly related in the literature to patients with hepatocarcinoma or cirrhosis.45 Stop codon substitution (C69*) was observed in the HBV genotype F sequence isolated in the present study with hepatitis Delta virus coinfection. This study participant had a history of advanced liver disease with a cirrhosis profile. This corroborates findings that correlate the C69* stop mutation with cirrhosis described by Veazjalali et al.46 in a homogeneous population infected with the HBV-D genotype.
In conclusion, the molecular characterization identified the HBV-D genotype as the most frequent in natives of the region, different from what other studies had shown, as the HBV-D genotype is frequently found in populations in the southern region of Brazil. The presence of vaccine escape mutations in the circulating HBsAg antigenic region may indicate changes in HBsAg antigenicity favoring HBV immune escape in the native population. In addition, the findings can contribute to the evaluation of immunization programs and assistance to vulnerable populations, favoring the prevention of viral hepatitis.
